Qu’est-ce que le syndrome d’Angelman ?
Vous vous posez la question : qu’est-ce que le syndrome d’Angelman ? alors vous êtes au bon endroit. Nous allons commencer par une définition puis établir les causes génétiques, son diagnostic et enfin évoquer les différentes formes épileptiques présentes dans le syndrome.
Le syndrome d’Angelman (SA) est un trouble neurogénétique rare qui affecte environ un enfant sur 15 000 – environ 500 000 personnes dans le monde.
Le SA est un trouble neuro-développemental important. Il est caractérisé par un retard sévère du développement avec une déficience intellectuelle importante, une absence de langage oral, des troubles de la motricité, de l’équilibre et de la sensorialité.
Certaines personnes ne marchent jamais. Les cycles de sommeil perturbés peuvent également être un défi sérieux pour l’individu et l’entourage. Les personnes atteintes du SA ont besoin de soins continus et sont incapables de vivre de manière autonome. Ils ont une espérance de vie normale. C’est la vie aujourd’hui des personnes atteintes du SA, mais l’espoir est là. Les scientifiques pensent que le SA a le plus grand potentiel de guérison par rapport à d’autres troubles neurogénétiques et FAST (Foundation Angelman Syndrome Therapeutics) a une feuille de route pour y parvenir.
Le nom de ce syndrome vient du Docteur Harry Angelman qui publia pour la première fois en 1965 un article de recherche décrivant cette maladie rare. Il a fallu plus de 20 ans aux chercheurs pour découvrir que l’anomalie génétique provenait du chromosome 15 et douze années supplémentaires pour isoler le gène responsable du syndrome : le gène UBE3A. L’identification du gène du SA en 1997 a complètement changé la donne pour la communauté Angelman.
La protéine UBE3A produite par le gène du même nom est nécessaire à la dégradation et au recyclage des protéines dans nos cellules dans un processus appelé l’ubiquitination. Chez toutes les personnes atteintes ou non du SA, seule la copie maternelle du gène UBE3A s’exprime dans le cerveau, la copie paternelle est silencieuse.
Caractéristiques
Les caractéristiques typiques du SA ne sont généralement pas visibles à la naissance. Les personnes atteintes présentent des difficultés d’alimentation dès l’enfance et un retard de développement notable vers l’âge de 6 à 12 mois. Ils ont besoin de rééducations intensives pour développer leurs compétences fonctionnelles. Le SA affecte toutes les races et les deux sexes. Il est souvent diagnostiqué à tort comme un autisme ou une paralysie cérébrale.
Comportements
Les personnes atteintes du syndrome d’Angelman ont des caractéristiques comportementales spécifiques, notamment un comportement heureux, caractérisé par des rires, des sourires et de l’excitabilité fréquente. Beaucoup de personnes atteintes du SA ont une fascination pour l’eau et prennent un grand plaisir dans des activités comme la natation et la baignade.
Il est important de noter que le syndrome d’Angelman est un trouble du spectre et qu’en tant que tel, tous les individus ne présentent pas les mêmes caractéristiques ou préférences comportementales.
Le syndrome d’Angelman est un trouble monogénique causé par la perte de fonction du gène UBE3A sur le 15ème chromosome maternel. Tous les individus ont deux ensembles de chromosomes – l’un hérité de la mère et l’autre du père. Le gène UBE3A hérité de la mère s’exprime, tandis que la copie du gène hérité du père est réduite au silence dans les neurones de notre cerveau – un phénomène connu sous le nom d’empreinte. Pour les personnes atteintes du SA, ce gène maternel ne fait pas son travail, et cela a un impact sur leur ARN messager (ARNm).

Qu’est-ce qu’ARNm ?
L’ARNm est le messager de notre corps. Notre ADN (ce qui constitue nos gènes) utilise l’ARNm en tant que service de livraison pour envoyer des consignes aux usines d’assemblage de protéines de nos cellules. Les personnes atteintes du SA ont une mutation, une délétion ou un autre défaut dans leur gène UBE3A, ce qui interrompt ce service de livraison. En conséquence, leurs neurones fabriquent peu ou pas de protéine UBE3A fonctionnelle, ce qui déclenche les symptômes du SA. Cette protéine est ce qui nous aide à marcher, parler et effectuer d’autres tâches quotidiennes.

Est-ce héréditaire ?
Dans la plupart des cas, le syndrome d’Angelman n’est pas hérité – en particulier ceux causés par une délétion ou une disomie uniparentale ; ces modifications génétiques se produisent de manière aléatoire au cours de la formation de cellules reproductrices ou dans les premières phases du développement embryonnaire.
Les génotypes
L’ADN (acide désoxyribonucléique) est le principal composant des chromosomes. Il contient notre code génétique unique. Le syndrome d’Angelman implique une perturbation du gène UBE3A sur le 15e chromosome maternel sur la région 15q11-13, l’expression du gène étant naturellement silencieuse sur le chromosome paternel. Différents tests sont nécessaires pour déterminer le génotype spécifique : délétion, mutation (UBE3A), disomie uniparentale (UPD) ou défaut d’impression (ICD). FAST poursuit activement des stratégies thérapeutiques pour tous les génotypes.
Plus d’informations sur les tests génétiques sont disponibles ici
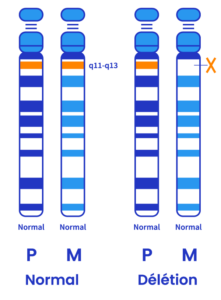
Délétion (entre 65 et 75%)
Pour la plupart des personnes atteintes du SA, il manque un morceau d’ADN sur le chromosome maternel et sur une portion plus ou moins longue incluant la suppression du gène UBE3A
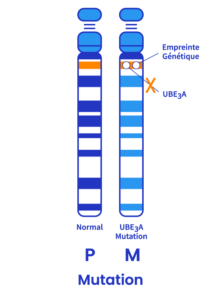
Mutation (5-11%)
Cela se produit lorsqu’il existe une petite anomalie dans l’ADN du gène UBE3A. Une mutation peut se produire n’importe où sur le gène.
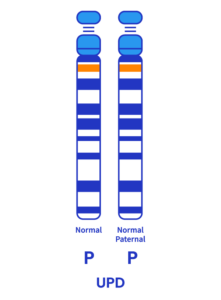 Disomie uniparentale (3-7%)
Disomie uniparentale (3-7%)
Un individu atteint de disomie uniparentale possède deux copies du chromosome 15 de son père, en lieu et place d’une copie provenant de sa mère et une autre de son père.
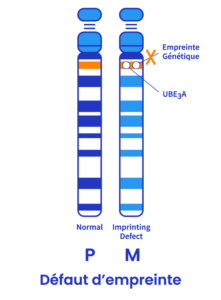
Défaut d’empreinte (<3%)
Le centre d’impression est une petite portion d’ADN situé dans la région q11-13 du chromosome. Dans de rares cas, le chromosome 15 de la mère est vierge et le centre copie le chromosome 15 du père. Il s’agit d’un défaut d’impression.
Dans certains cas, la cause génétique n’est pas clairement identifiée et le diagnostic s’établit dans le cas présent sur les signes cliniques.
Le diagnostic est avant tout clinique devant les retards du développement de l’enfant constaté par un médecin / pédiatre.
Une consultation dans un centre génétique est nécessaire pour rechercher les anomalies génétiques et permettre aussi le conseil génétique.

Signes cliniques
Le cheminement vers le diagnostic du syndrome d’Angelman n’est pas toujours facile. Il n’y a pas d’âge définitif pour le diagnostic, mais grâce aux progrès scientifiques et aux dépistages chez les nourrissons, le diagnostic se fait de plus en plus tôt. Les signes cliniques du syndrome d’Angelman varient en fonction de l’individu et du génotype.

Tests génétiques
Il existe différents types de tests pour déterminer si votre enfant est atteint du syndrome d’Angelman. Nous fournissons des informations aux parents et aux personnes qui n’ont pas une connaissance pratique de la génétique, mais qui aimeraient comprendre quels tests sont disponibles et ce qu’ils signifient.
Malheureusement, l’épilepsie est courante chez les personnes atteintes du syndrome d’Angelman (SA). Environ 85% des personnes atteintes du SA connaîtront des crises au cours des trois premières années de leur vie, bien que les crises dans le SA puissent se présenter à tout âge. La bonne nouvelle est qu’il existe de multiples options de traitement efficaces pour gérer les crises chez les patients atteints du SA, notamment des médicaments antiépileptiques, une intervention diététique thérapeutique ou encore un implant de stimulateur du nerf vagal.
Que vous soyez nouvellement diagnostiqué ou que vous ayez le diagnostic depuis des années, être en mesure de reconnaître une crise, de communiquer correctement les symptômes à l’entourage de votre enfant et de connaître les meilleures pratiques pour traiter les crises d’épilepsie sera votre meilleure défense pour assurer la sécurité et la santé de votre enfant.
Les personnes atteintes de SA peuvent présenter différents types de crises, notamment :
- Absence atypique
- Généralisé tonico-clonique
- Atonique
- Myoclonique
- Focal
- État de mal épileptique non convulsif
Si votre enfant a récemment reçu le diagnostic du SA, parlez à votre médecin de l’implication d’un neurologue s’il ne le fait pas de lui-même, afin d’envisager la planification d’un EEG (électroencéphalogramme) de base. Le neurologue sera en mesure de mettre en place les traitements médicamenteux éventuels pour contrôler au mieux les crises.